Avoiding the Pitfalls of PQR

Summary
Annual Product Quality Reviews (APQRs) are required to confirm process consistency, specification fit, and the need for CAPAs, but many teams struggle with manual collection, fragmented systems, and slow reporting.
The paper shows how Continued Process Verification (CPV), supported by integrated data sources, can streamline APQR inputs and move teams toward a more timely “real-time APQR” approach.
Key takeaways
- APQRs check process “state of control,” validation status, specification suitability, and trends that may drive CAPAs or even revalidation.
- Common APQR pitfalls include disconnected data sources, audit-readiness delays, evolving multi-region requirements, weak data governance (ALCOA+), and manual, inconsistent reporting.
- A CPV program (Stage 3 expectation) can source key datasets directly and, with MES/ERP/LIMS plus CAPA/change control integration, support more timely APQR reporting and analysis.
Who is this for
- Quality Assurance (QA) managers and QA/QMS leads
- Quality Control (QC) managers and laboratory leaders
- CPV / process monitoring leads and analysts
- Process engineers / manufacturing science & technology (MS&T) professionals
- Validation / qualification and CSV professionals (equipment and computerized systems)
- Regulatory affairs professionals supporting post-approval and filings alignment
- Data integrity and compliance specialists (ALCOA+ governance)
Download your Industry Insight
Avoiding the Pitfalls of APQR
The Annual Product Quality Review (APQR) is a regulatory requirement designed to ensure consistent product quality across the pharmaceutical product lifecycle. While foundational to GMP compliance, traditional APQR practices—characterized by manual data collection, fragmented systems, and delayed reporting—often struggle to meet the dynamic needs of modern quality systems.
This Industry Insight explores the typical challenges organizations face in executing effective APQRs, including data integrity risks, regulatory complexity, and inefficient reporting processes. It highlights the growing need for innovation and digitalization to transform APQR from a static, compliance-driven task into a proactive quality management tool.
A key enabler in this transformation is Continued Process Verification (CPV) programs, which provide real-time process and quality data through integration with external data systems. By adopting a digital CPV backbone, organizations can streamline APQR workflows, reduce operational burden, and enable timely, data-driven insights.
Ultimately, a digital transformation approach that combines technological integration with empowered teams is essential to support the shift toward a “real-time APQR” model—improving compliance, operational efficiency, and continuous quality improvement.
Introduction
The annual product quality review is an essential topic for pharmaceutical and biopharmaceutical manufacturers, serving as a crucial mechanism to demonstrate product quality and compliance in a centralized fashion.
According to the U.S. Food and Drug Administration (FDA), the APQR is a regulatory expectation and should be conducted at least once per year (FDA, 2024). While other regulatory authorities, such as the European Medicines Agency (EMA) do not explicitly use the term “annual” or “APQR,” the practice of performing a yearly product quality review is widely adopted across the EU regulatory framework (European Commission, 2013). The frequency of the APQR may be adjusted based on factors such as product complexity, manufacturing volume, and historical process performance, but as a general rule, it is recommended to conduct the review at least annually to ensure continued product quality and compliance.
Despite its frequency and the size of the product portfolio, the process of creating an APQR can be cumbersome—a journey paved with inefficiencies and data integrity risks, from data collection to analysis, interpretation, and finally, reporting.
In this Industry Insight, we will explore strategies to streamline these labor-intensive tasks and go beyond a mere “check in the box” for regulatory compliance. We begin with APQR fundamentals and then describe the typical challenges manufacturers face in meeting APQR’s primary goals. Later, we present solutions aimed at unleashing the potential of a “real-time APQR” built on a digital CPV program, founded on a proactive, deliberate approach to product quality and continuous improvement.
By investing in an efficient workflow supported by a digital backbone operating under a single platform, subject matter experts (SMEs) can redirect their focus toward more value-added tasks and seize opportunities for process and quality enhancements.
Purpose and Regulatory Landscape of APQR
In simple terms, APQR is an evaluation conducted to assess the quality standard of each product:
- Verify the consistency of the existing process, “state of control,” and validation status
- Check the appropriateness of current product specifications
- Analyze any trend to determine the need of corrective actions and preventive actions (CAPAs), which may include changes to specifications, manufacturing processes, and/or adjustments to the control strategy
Regulatory agencies such as the FDA 2024, require manufacturers to conduct an annual review of drug product quality to determine whether changes are warranted in product specifications, manufacturing processes, or control procedures.
Similarly, the European Commission describes the Product Quality Review (PQR) as a tool to verify the consistency of the existing process and the appropriateness of current specifications for both starting materials and finished products. The objective is to highlight any trends and identify product and process improvements, thereby contributing to an effective control strategy.
Within the framework of internationally recognized quality standards—such as ICH Q7A (International Conference on Harmonisation, 2000), ICH Q10 (International Council for Harmonisation, 2008), and PI 009-17 Part I (Pharmaceutical Inspection Co-operation Scheme (PIC/S), 2023)—APQR is considered an essential element of the pharmaceutical quality system. While not specifically defined as “APQR”, the concept aligns with the product quality review principles outlined in these guidelines, which emphasize ongoing verification of a state of control.
The APQR serves as a diagnostic tool—akin to an “X-ray” of the product and process—that helps determine whether CAPAs are needed. Under certain conditions, a CAPA may warrant process revalidation. For example, a review of change controls may reveal that several incremental changes collectively threaten the validated state, or the presence of recurring out-of-trend (OOT) or out-of-specification (OOS) results may indicate that the process is not in the desired state of control.
APQR Goals
In addition to meeting regulatory requirements, APQR supports broader organizational goals that drive quality and business excellence.
Quality Right First Time
Reduce or eliminate OOS results that could have been avoided through early identification of deviations in process or product behavior from the validated state of control. Enable the identification of opportunities for continuous improvement that go beyond compliance with product specifications.
Harmonization
Standardize procedures, templates, and systems across the organization to ensure consistency and uniformity in the execution of APQRs, enabling reliable cross-product comparability and facilitating organization-wide quality oversight.
Scalability
Enable efficient handling of increasing data volumes as the organization grows, without compromising performance, accuracy, or the integrity of the APQR process.
Cost Reduction
Effectively manage costs associated with implementing, maintaining and operating APQR processes while ensuring high standards of quality and compliance. From a manufacturing process standpoint, actively pursue cost optimization by exploring process improvement opportunities, such as enhancing yield, purity, and throughput.
APQR Challenges
The journey toward achieving these goals can be hindered by the following challenges:
Reactive Approach to Quality
Without a robust trend analysis framework, emerging deviations may go undetected, increasing the risk of quality issues and missed opportunities for continuous improvement. Inadequate data trending also undermines the effectiveness of root cause investigations, leading to suboptimal or misaligned CAPAs.
Regulatory Compliance
Organizations often face several challenges in ensuring regulatory compliance throughout the APQR process, including:
- Multiple information sources: The use of disparate systems—such as spreadsheets, and paper records—makes it difficult to consolidate data effectively, potentially leading to gaps or oversight in APQR documentation.
- Audit readiness: Lengthy APQR workflows can delay report preparation, which may increase the risk of noncompliance during audits or inspections.
- Evolving regulatory requirements: Regulatory landscapes continue to evolve, requiring organizations to frequently adapt to new or updated regulatory expectations. Maintaining thorough, well-structured documentation that satisfies requirements across different jurisdictions adds complexity and can hinder the development of a harmonized, globally compliant APQR process.
Data Management
Because the APQR process involves the collection and analysis of a comprehensive data package, organizations often encounter several common challenges:
- Data collection: One of the most common pitfalls in the APQR process is the failure to collect comprehensive and accurate process and quality data. This often results from incomplete or missing data and inconsistent or unstandardized data sources, which can hinder effective analysis and decision-making.
- Data accessibility: A lack of a centralized data repository—caused by disconnected systems and fragmented tools—prevents the APQR process from being fully collaborative and traceable. Without integrated access, it becomes difficult to ensure secure, controlled access to data and maintain data integrity, both critical for regulatory compliance.
- Data integrity: Ensuring data integrity is essential to the APQR process, as all data must be accurate, consistent, and reliable. Implementing robust data governance and management practices, including access controls, audit trails, and standardized procedures, is necessary to uphold ALCOA+ principles and ensure regulatory compliance.
Reporting
Effective APQR reporting relies on streamlined processes, standardized templates, and effective data compilation. Key challenges include:
- Manual and time-consuming reporting: APQR report generation often involves manual collection, collation, and formatting of data and charts from multiple sources. This process consumes significant resources and time and increases the risk of errors and inconsistencies.
- Lack of standardized processes and templates: The use of varied formats and site-specific templates across products makes it difficult to standardize APQR outputs and identify systemic trends. These inconsistencies hinder the ability to derive meaningful organization-wide insights from APQRs.
In the next section, we will cover solutions to help tackle these challenges.
CPV as a Key Enabler of APQR
The APQR covers specific cGMP-related elements associated with the product lifecycle, including (but not limited to): starting and packaging materials and suppliers; in-process control (IPC) and final product (FP) testing data; OOS/OOT results, deviations, investigations, and associated CAPAs; stability data and trend analysis; change controls and their impact on product quality, equipment qualification and maintenance data; product recalls, complaints, and returns; and evaluation of GMP compliance of outsourced activities. From a regulatory affairs perspective, the APQR should also include a review of marketing authorization variations (submitted, approved or rejected) across different regulatory jurisdictions to ensure alignment between manufacturing practices and regulatory filings.
While some APQR sections may remain relatively stable over a defined period, others related to batch manufacturing tend to evolve more rapidly as new process and quality data becomes available. In the era of Pharma 4.0 and rapid technological advancement, managing the lifecycle of large volumes of data presents significant challenges, especially when trying to achieve operational efficiency at scale—such as when dealing with extensive product portfolios manufactured at different sites and across geographies.
The figure below illustrates the typical sections of an APQR where the most time-consuming tasks reside.
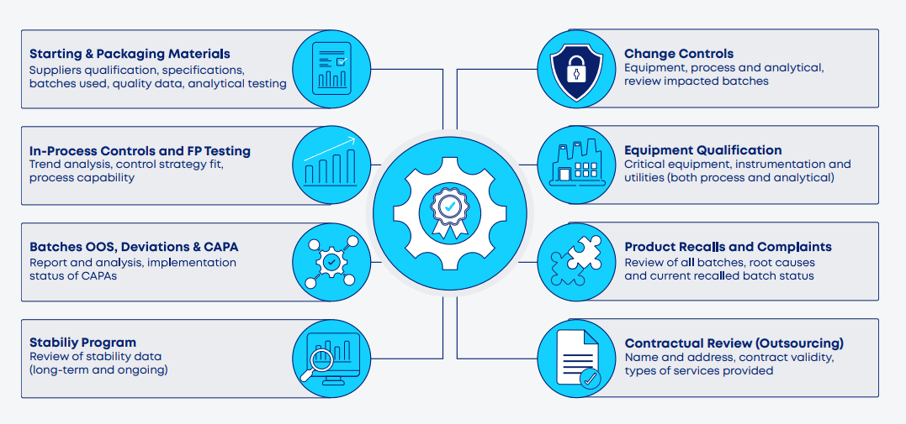
Figure 1. Main sections of a typical APQR, with emphasis on where most of the effort resides for data collection, analysis, and reporting.
This is where an APQR, strengthened by a robust and efficient CPV program, can elevate both quality and business efficiency to the next level.
According to the FDA guidance for process validation 2011, CPV is a regulatory expectation in Stage 3 of the product lifecycle, which focuses on “…continual assurance that the process remains in a state of control (the validated state) during commercial manufacture....” This means that, through CPV, manufacturers are expected to continuously monitor process performance and product quality to ensure ongoing control and proactively identify areas of improvement.
In essence, CPV can significantly streamline the collection and analysis of periodic data for the APQR, as key datasets, such as product quality, process performance, and stability data, can be directly sourced from the CPV program.
Therefore, typical challenges such as a reactive approach to quality, inefficient data management, data integrity concerns, and labor-intensive reporting can be effectively mitigated through a well-implemented CPV program. A digital CPV system further enhances reporting capabilities at predefined time intervals. This not only improves consistency but also frees up valuable bandwidth for technical teams to focus on higher-value activities, such as conducting root cause analysis of process deviations that require attention.
A complete review of the qualification status of critical equipment, instrumentation, utilities, and computerized systems supporting cGMP activities (both process and analytical) is also a mandatory component of the APQR. Digital platforms that manage the full lifecycle of equipment and computerized validation systems play a key role in ensuring compliance, enhancing data availability, and improving operational efficiency. By leveraging such systems, organizations can ensure that up-to-date asset qualification and inventory status are readily accessible from any location, thereby streamlining APQR preparation and enabling timely, data-driven reporting.
Unlock the Potential of APQR
To fully unlock the potential of a “real-time APQR” enabled by a digital CPV program, organizations should embark on a digital transformation that focuses not only on technology adoption but also on empowering people as key enablers of change.
While digital roadmaps may vary in ambition and maturity, external drivers are pushing organizations toward Industry 4.0. These include regulatory expectations, the need to reduce operational costs and accelerate time to market, the rise of advanced technologies (e.g., data analytics, automation, interconnected systems), and growing product and process complexity characterized by high volumes of process and quality data.
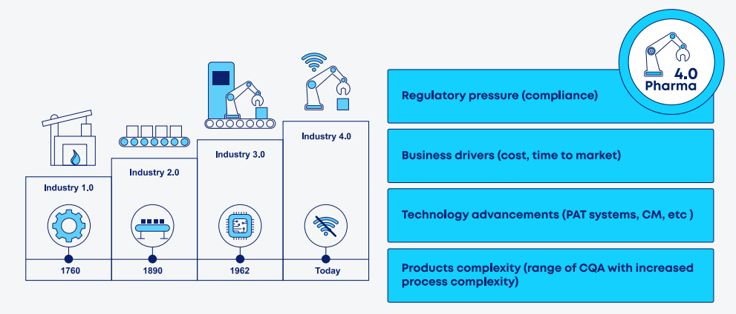
Figure 2. Main drivers pushing organizations toward digital transformation.
As shown in Figure 3, a schematic representation of the data sources and system integrations that form the foundation for achieving a “real-time APQR.” Here, all relevant data sources required to support the CPV program (including both process and quality data) are funneled into an aggregated and centralized database. Real-time data acquisition—pivotal for an automated CPV program—is enabled through the seamless integration of multiple systems, such as manufacturing execution systems (MES), enterprise resource planning (ERP), and laboratory information management systems (LIMS). This integration allows for real-time data collection and storage.
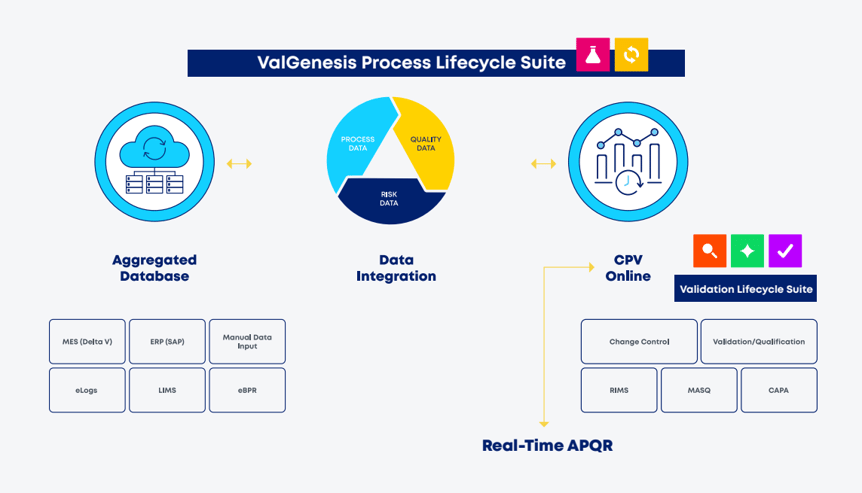
Figure 3 - Schematic representation of data sources and system integrations to achieve a “real-time APQR.”
Furthermore, connecting with quality systems such as CAPA and change control further enriches the dataset and supports a dynamic, compliant, and efficient “real-time APQR.”
Conclusion
The APQR is a multidimensional regulatory requirement designed to ensure product quality consistency across the entire product lifecycle. However, it is increasingly recognized that traditional, discrete APQRs that offer only a point-in-time view are no longer sufficient to meet the evolving demands of modern quality systems and business operations.
Addressing the challenges of APQR requires innovative, technology-driven solutions to uphold product quality and ensure ongoing GMP compliance. By leveraging digital infrastructure and integrated systems to enable comprehensive automation, pharmaceutical manufacturers can transition from traditional APQR processes toward a more agile, “real-time APQR” model.
Investing in a streamlined, digitally-enabled workflow empowers organizations to build both business and quality resilience while also unlocking operational efficiencies. This shift frees up technical teams from manual tasks, allowing them to focus on higher-value activities such as root cause analysis, process optimization, and proactive quality improvements.
References
- European Commission. (2013). EudraLex Volume 4, Chapter 1: Pharmaceutical Quality System. The Rules Governing Medicinal Products in the European Union. Retrieved from https://health.ec.europa.eu/document/download/e458c423-f564-4171-b344-030a461c567f_en?filename=vol4-chap1_2013-01_en.pdf
- International Conference on Harmonisation. (2000). ICH Q7A: Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients. Retrieved from https://www.ema.europa.eu/en/ich-q7-good-manufacturing-practice-active-pharmaceutical-ingredients-scientific-guideline
- International Council for Harmonisation. (2008). ICH Q10: Pharmaceutical Quality System. Retrieved from https://www.ema.europa.eu/en/ich-q10-pharmaceutical-quality-system-scientific-guideline
- Lighthouse. (2023). The History Of Pharmaceutical Manufacturing: Industry 1.0 to Pharma 4.0. Retrieved from https://www.golighthouse.com/en/knowledge-center/industry-1-pharma-4/
- Pharmaceutical Inspection Co-operation Scheme (PIC/S). (2023). PI 009-17 Part I: Guide to Good Manufacturing Practice for Medicinal Products. Retrieved from https://picscheme.org/docview/6606
- U.S. Food and Drug Administration. (2011). Guidance for Industry: Process Validation – General Principles and Practices. Retrieved from https://www.fda.gov/files/drugs/published/Process-Validation--General-Principles-and-Practices.pdf
- U.S. Food and Drug Administration. (2024). Code of Federal Regulations Title 21, Part 211: Current Good Manufacturing Practice for Finished Pharmaceuticals. Retrieved from https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211