
Abstract
Cleaning validation (CV) is a critical requirement in pharmaceutical manufacturing, ensuring patient safety, product quality, and regulatory compliance. Yet despite its importance, CV is often deprioritized and managed through legacy, paper-based approaches that are inefficient, error-prone, and resource intensive. These outdated practices reinforce the perception of CV as a burden rather than an enabler of operational excellence.
This Industry Insight explores how digitalization transforms cleaning validation from a compliance checkbox into a strategic driver of efficiency, adaptability, and continuous improvement. It examines the regulatory expectations set by agencies such as the FDA, EMA, and the World Health Organization (WHO), all of which emphasize science- and risk-based lifecycle approaches. It also outlines how digitalized systems enable audit readiness, knowledge management, and consistent application of best practices across global networks.
Key benefits of digital cleaning validation include streamlined execution, reduced downtime, improved data integrity, and enhanced traceability. Integration with enterprise platforms such as LIMS, MES, and ERP further ensures lifecycle continuity, while scalability supports both centralized oversight and local flexibility. Looking ahead, the adoption of Pharma 4.0 principles and artificial intelligence (AI) offers opportunities for predictive insights, continuously optimized cleaning cycles, and adaptive validation strategies.
By modernizing cleaning validation with digital tools, life sciences organizations can strengthen compliance, unlock operational agility, and accelerate time-to-market while maintaining the highest standards of patient safety.
Introduction
Cleaning validation (CV) is a critical element of compliance, patient safety, and operational efficiency in pharmaceutical manufacturing. While the compliance and patient safety elements are easy to recognize, they are not so easy to achieve. Despite advances in digitalization across qualification and validation lifecycles, CV often remains reliant on spreadsheets, PDFs, and paper-based workflows. As a result, it is generally regarded as a “necessary evil” and “lowest priority” compared to other elements of a new process or product introduction, or in the startup of a new greenfield facility and the first full-scale introduction of manufacturing on new equipment.
In both circumstances, the requirement to complete cleaning validation is viewed as something to be done with low-resource intensity, minimal development, and a “nuclear” approach to ensure passing results are achieved the first time. Thus, any allowance for CV in a project is often the first element cut when a schedule crash is required, either because of delays in other areas or stakeholder urgency to achieve market release. In neither case are the resultant suboptimal outcomes of the cleaning validation exercise the fault of the CV team. Nor do they reflect a lack of desire to serve as operational enablers rather than hindrances. Instead, these outcomes stem from a systemic perspective that cleaning is a zero-sum game that consumes overall equipment effectiveness (OEE), rather than an opportunity — with minimal investment — to free significant percentages of OEE in new or existing manufacturing paradigms. In the following sections, we examine the factors that contribute to these perceptions and explore how digitalization can transform cleaning validation into a competitive advantage.
What Drives the Perception of Cleaning Validation as a Burden
Legacy paper-based approaches are poorly defined, time-consuming, error-prone, and ill-suited to today’s regulatory and operational demands. In most cases, these approaches burden CV and Operations with slow, exception-inducing processes that take significant time to prepare, pre-approve, complete, and post-approve. A less noted, but more detrimental impact, is that it is also a root cause of the inability to observe, learn, react, update, and maintain a constant state of readiness and cleaning process improvement agility. These preparation delays stymie the ability of cleaning validation subject matter experts (SMEs) to achieve efficient, optimized cycles across manufacturing processes and force execution teams to apply the maximal “nuclear” approach of “hit it with everything we’ve got because we can’t afford a failure.” This ingrains inefficiency, and therefore OEE consumption, into the system. The inability to be agile, learn fast, and execute without error ensures that senior stakeholders continue to block attempts to experiment or explore process limits. Instead, they accept ongoing OEE losses rather than weeks or months of delay in the race to market.
Modern, fully digitalized CV is not just a technology or process upgrade — it is a strategic shift that directly impacts batch throughput, inspection readiness, and scalability. Purpose-built, industry-design-led solutions can eliminate compliance gaps, reduce equipment downtime, and align CV processes with Pharma 4.0 principles.
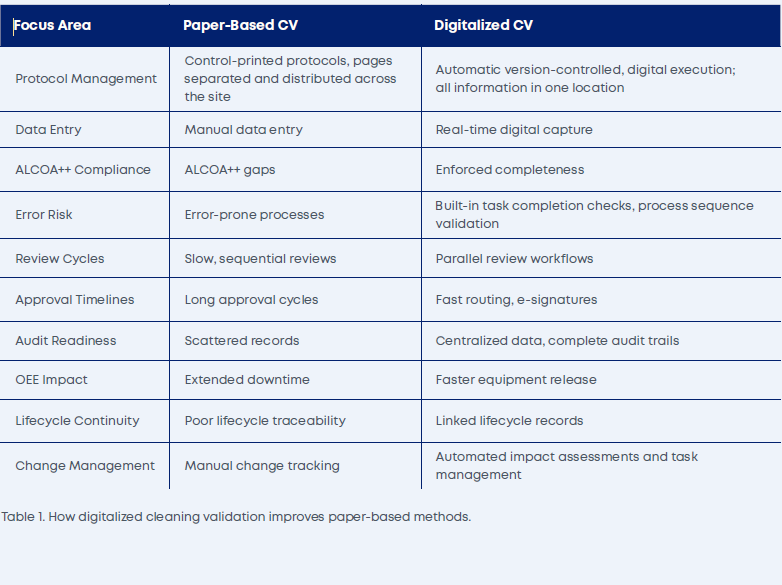
The contrast becomes clear when comparing traditional paper-based approaches with a modern digitalized approach, as shown in Table 1.
Compliance-Focused Development
Regulators such as the FDA, EMA, and WHO expect cleaning validation to be documented, traceable, and aligned with a risk-based lifecycle approach (FDA, 2011; EMA, 2015; WHO, 2014). Manual or semi-automated methods frequently fall short, creating gaps in traceability and version control, outdated protocols lost in paper format throughout a facility, and procedures that struggle to adapt to changing regulatory needs or an organization’s global guidance and expectations.
Documented Science- and Risk-Based Approaches
Lifecycle expectations begin with the assumption that manufacturers will have a science-based logic for setting cleaning parameters and selecting cleaning agents. This typically includes the completion of cleaning studies, often performed on coupons of different materials of construction at lab scale, supported by risk-assessment processes that examine the process intermediate composition, processing criteria, and potential cleaning agent characteristics and interactions with both equipment and process intermediates. The results of these risk assessments and studies should be directly traceable to the design of the cleaning process to support full lifecycle management. Digitalized risk assessment and study management allow for the direct linkage of these items to specific cleaning process parameters, stages, sequencing, and monitoring criteria (EU, 2015; ICH, 2005).
Worst-Case Rationale
Appropriate and detailed investigation of scientific properties — beyond a simple solubility criterion, which often lacks a minimal time-temperature-dissolution rate surface response understanding — should drive robust and constantly challenged worst-case product understanding. Data-backed business rules should be agreed upon and rigorously implemented across a program to ensure consistency of approach and outcome. This rigor should also be extended to equipment worst-case determination and documentation, with consistency across a network or business unit. At the same time, site-embedded SMEs should be able to incorporate experiential knowledge into sampling plan development above “standard” location requirements.
Plan for Audit-Readiness From Day One
A digitalized approach to cleaning program development helps mitigate common findings from inspectorates. Insufficient development activities and/or documentation of cleaning process development — and the rationale for translating this information into a robust, repeatable, and defensible science-based cleaning process — are avoided through system-mandated actions and risk assessments. Similarly, specific selection of residue limits and their rationales are often not documented adequately, which can also be avoided with system-mandated processes.
At a more fundamental level, a digitalized process ensures that data integrity deficiencies, such as missing audit trails or incomplete metadata, are avoided, and that electronic signatures are fully traceable across all records. Misalignment between corporate policies and site-level execution can also be avoided by applying frameworks for cleaning validation programs that ensure task activation and outcome achievement, extending through the full lifecycle (FDA, 2018; EU, 2015).
Table 1. How digitalized cleaning validation improves paper-based methods.
ALCOA++ Native Rapid Digital Execution
At-scale execution of cleaning process qualification and validation is one of the most time- and resource-consuming activities a manufacturing site must complete, which can be, perhaps unfairly, considered a non-value-added activity. It is a regulatory imperative that cleaning validation is completed, and with planning, technological support, and execution windows protected by sponsors, it can enable program executions to immediately release unexpected OEE once operations commence and unlock efficiency in legacy programs.
Rapid Execution, Every Time
Legacy cleaning validation approaches, heavily paper-based, inhibit the ability to rapidly execute and “move on” from any CV activity, qualification, or validation. Protocols must be control-printed, tracked, manually completed, and then recollected with evidence for roundtable review (if the process runs smoothly!) and sequential approval. This is slow, prone to data-entry errors, and incredibly complex to manage due to the sheer number of protocols, sampling plans, data tables, evidence printouts, result reports, and related records, which must be collected, maintained as a package, and handled by many resources in different business functions. Inevitably, items go missing or are temporarily lost, reviews stall, investigations stumble, approvals lag, and equipment and overall execution processes consume the allotted schedule window before all of the necessary work has been completed.
Fully digitalized execution eliminates almost all of these challenges. Digitalized protocols and plans are instantly accessible at all times, even offline on the manufacturing floor during challenging facility startups. Entry errors, missing signatures, unreadable data, and other ALCOA++ issues are not possible, particularly where integrations with MES/DCS or other equipment and component systems have been achieved, and data and information can be captured in real time, evidence attached instantly and permanently, and field-level controls enforce full and correct completion before task closure. At the same time, these activities are recorded in an always-on, immutable audit trail and governed by Part 11/Annex 11 controls (e.g., enforced completion rules, controlled templates, validated calculations, and electronic signatures) so that electronic records and electronic signatures (ERES) and data integrity requirements are a matter of course, and not a source of anxiety (FDA, 2018; FDA, 1997; EMA, 2011).
New Program Efficiency From the Start
How do all of these digitalization wonders impact site operational efficiency for a greenfield site? To begin with, cleaning validation execution with this level of control and integrated error reduction ensures each iteration of a cleaning cycle and testing is completed on time, every time. Additionally, the corresponding review and approval activities can be initiated, and the workflow can be completed as soon as the executor completes the last item and submits the activity into the system. Potentially, digitalization allows review, and even approval, to be completed before the executor has degowned out of production to go for a well-deserved coffee or to submit swab samples to QC.
Execution efficiency and accuracy at this level enable project management to have confidence in the resource requests, facility time estimates, and outcomes the cleaning validation team wishes to achieve. This empowers all parties, particularly Operations and Manufacturing stakeholders, to request and achieve the cleaning process optimization they desire for plant efficiency — but which they previously worried (with just cause) would consume inordinate time, resources, and money. Thus, from day one of production, operational teams have a world-class turnaround and cleaning process without the world-class headache.
Legacy Program OEE Optimization Without the Panic
Legacy cleaning programs almost universally suffer from the inefficiencies inherent in over-cleaning or “nuclear” cleaning, driven by rational fears and implications of the challenges discussed above. The reasonable approach, when historically facing this fleet of unknowns and opportunities for failure, was to reduce the overall number of cycles to be qualified, increase the breadth of product and equipment groups, and design a cleaning process guaranteed (+150%) to clean the worst of the worst and apply it to all. This is a successful and robust approach to achieving cleaning requirements, but it also ensures that cleaning will consume inordinate amounts of operational time indefinitely.
The challenge with the “over-clean” approach from historical cleaning processes and validation programs is that it is almost impossible to convince stakeholders to engage in the retrospective analysis, cleaning validation re-executions, and support studies necessary to reclaim lost OEE. Why? For all of the issues discussed previously that are problematic with traditional, non-digital approaches. Documentation, lab studies, risk assessments, development runs, verification exercises, and actual cleaning validation execution are still laborious, error-prone, and potentially even more likely to go off schedule in a live GxP facility, as the knowledge and skills needed to deliver new cleaning validation activities may be rusty.
Furthermore, from a stakeholder perspective, the facility is in live production and has existing supply chain planning commitments for the next six to 18 months. Slowing down batch takt rate, risking validation batches not being available for market release, and potentially stopping all production activities for several weeks or months is seen as a greater danger to market supply than the long-term benefits. Even where the benefits are modeled to be significant, they are always couched with significant risk elements. Thus, facilities will trundle along for years, underperforming without fully understanding or examining the alternative — until their productivity is challenged by a new facility in the network or a new CDMO service provider, and the viability of the legacy site looks questionable.
The magnitude of these losses becomes evident when examining time-loss patterns across the lower performing facilities, as shown in Figures 1 and 2.
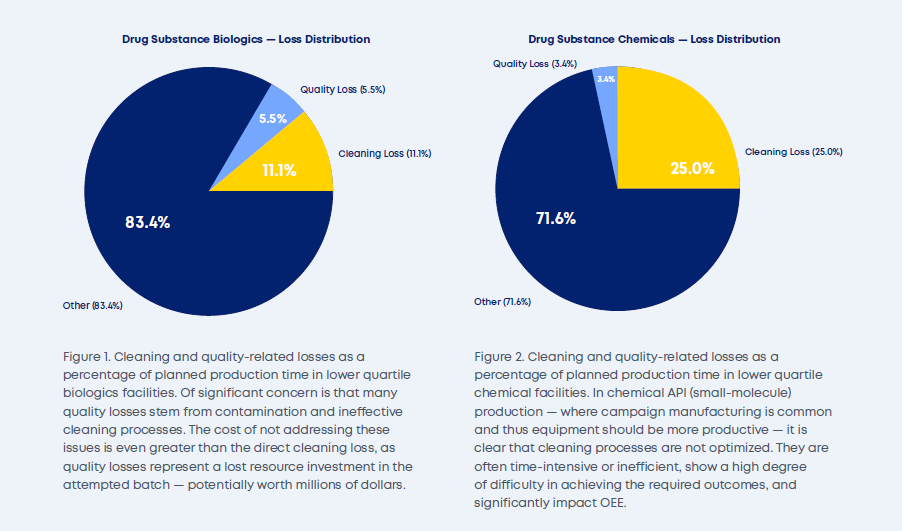
This is exactly the scenario where digitalized cleaning validation can provide a significant remedy, empowering legacy cleaning validation programs to strive for optimization and continuous improvement.
Digitalization Can Unlock Your Cleaning Validation
How can a digital tool overcome these historical issues? Firstly, the most precious commodity in a legacy facility and cleaning program is time. Time lost means batches are not produced. Digitalized validation processes continuously enable rapid, real-time delivery of validation by releasing equipment quickly for the next task with instant updates of activity completion, accelerating review and approval cycles by instantly placing completed activities into the to-do list of workflow participants, escalating issues more rapidly and with better baseline information to speed up investigation and remediation, controlling document spread and losses across a facility, and driving consistency of execution. They also enable database documentation of “tribal knowledge” in notes, justifications, assessments, and investigations that are key to success.
Finally, digitalized systems enable smaller SME teams to achieve better outcomes with diverse operations groups by driving consistency of information, onboarding and training, process execution, and data collection for analysis. User-level alerts and task management help prevent gaps in execution. This is supported at a stakeholder level with centralized, robust, real-time visibility at both a granular and program level.
Ongoing Verification and CV Program Lifecycle Management
The last (and first) phase of the cleaning program lifecycle is the ongoing verification and optimization stage. In this stage, the existing validation environment must be maintained and monitored, and the data collected over time should be analyzed to determine opportunities for optimization and efficiency, while also ensuring that drift or deviation from the validated design space is avoided and product quality is maintained (FDA, 2011).
Enterprise Integration and Knowledge Management
Integration with LIMS, MES, and ERP systems in any system is critical to ensure total lifecycle management. This should allow data to be pushed or pulled from one business-critical system to another, ensuring data structure and integrity are maintained. The data can take the form of individual device measurement values, tables of historical data, or action triggers for potential out-of-specification results or equipment status changes. The integrations that are possible in digitalized systems are also crucial enablers for total lifecycle knowledge management. It is only with connected databases that knowledge management can be realized and advanced.
Change Management
In addition to managing knowledge, the key challenge of phase 3 — and the essential kickoff point to return to phase 1 again for further development — is change management. With digitalized systems, the process of risk assessment, impact assessment, change management, and program updates is streamlined. Individual risks can be reviewed and updated, potential changes and impacts can be tested in sandbox environments, with immediate updates of residual limit calculations and worst-case strategy impacts — all without hours of manual spreadsheet review or paper review of existing cleaning processes and complex multi-process, multi-equipment worst-case logic. The risks posed by spreadsheets (validated or not) and manual calculation checks are permanently removed from the cleaning program.
Eternally Audit-Ready
In the era of digitizing and digitalization, one thing above all others can be singled out as a primary requirement, a quality prerequisite and enabler, and an audit preparedness must-have: audit trails. With advanced digitalization tools, organizations gain the assurance that every operator action, every submitted piece of data, every justification or observation, and every subsequent change and update are fully audit-trailed in compliance with regulatory best practices. The who, what, when, where, and why of your cleaning program are continuously tracked and immutably recorded. Thus, whether for internal or external purposes, organizations can have confidence that their cleaning programs are always inspection ready.
Scalability and Standardization
A final key to the success of any digital solution for a life sciences organization is scalability. What does this mean in practice? It means that global thought leaders and SME teams can shape the validation program for the entire organization through a single system. This single system enables multiple sites to be established and multiple validation programs within a site, all organized under a global business unit framework that matches the operational paradigm. Within this structure, standardized templates, frameworks, business rules, and procedures can be rolled out, driving consistency and reliability of execution while keeping costs and timelines streamlined.
Critically, standardization must also allow the other element of scaling — local flexibility. While procedures and frameworks can be driven from a global perspective, each site and each program must have certain degrees of freedom and customization that they can undertake with documented justification. Thus, the uniqueness inherent in every organization can be accommodated, and the need for continuous gap assessment against global procedures can be avoided. Centralized oversight will always be able to immediately see where the differences are, and where necessary, be remotely involved in the ongoing review, approval, and maintenance (or removal) of these differences.
Scalability and standardization should be further enhanced with system-wide access to a shared knowledge database. This can apply to risk assessments, documents, cleaning processes, or impact assessments. Data migration strategies from legacy records can also be achieved. This centralized shared knowledge, and the ability for any program to inherit or utilize information or output from another program, greatly empowers teams to reduce variability and shorten validation times. The collective outcome is that equipment, processes, and facilities onboard faster, achieve mobilization and validation in a lean but optimized manner, and reduce product time-to-market.
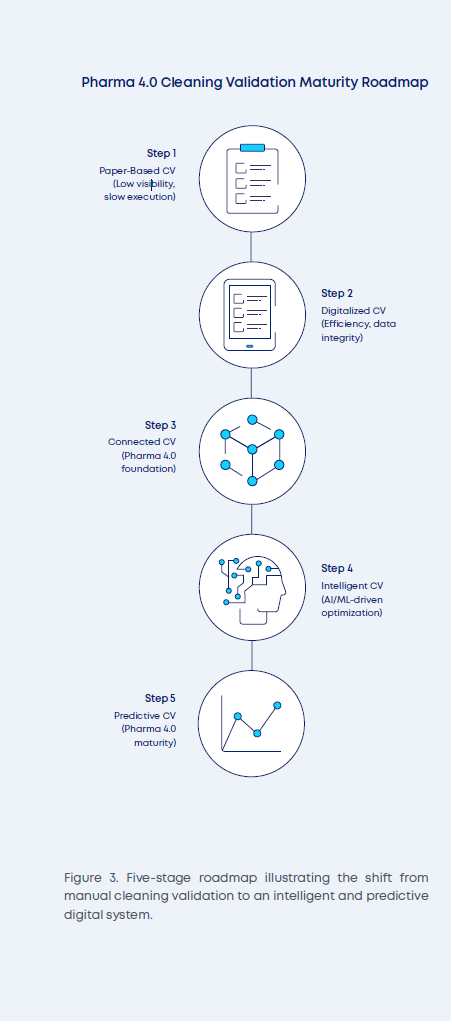
Pharma 4.0 and AI Integration
The digitalization of a cleaning validation program lays the foundation for an ongoing industry revolution: Pharma 4.0 and AI Integration (see Figure 3). Pharma 4.0 is gradually moving facilities toward operating modalities where data and control systems are fully connected and integrated. This can lead to further validation benefits by enabling the collection of required equipment and process data into the validation system at the touch of a button, without the risk of transcription errors (ISPE, 2023).
The major benefit, however, is likely to be in ongoing program maintenance and optimization. Without prompting, the digital CV system will continuously compile cleaning performance information, provide defined interval reports, and critically enable increasingly complex monitoring criteria to be established to ensure consistent outcomes. With the addition of machine learning, these systems will also be able to identify when baselines have shifted, when process capability has narrowed, or potentially predict when maintenance or verification activities may be required due to an increasing accumulation of variability or out-of-trend results, which may not necessarily trigger a request for review until they become out-of-specification results.
As AI capabilities increase, and as we purposefully train models on actual production data, it will be possible to use AI to examine the endpoints and outcomes of cleaning and recommend cycle adjustments. These adjustments can support compliance and efficiency by indicating where endpoints may be consistently close to the edge of failure, or where the endpoint is consistently achieved well in advance. Thus, safeguarding or optimizing opportunities can be flagged without the effort of manual data compilation and review, and SME resources can be focused on areas where opportunities have already been pre-screened, and meaningful impact is likely.
The same data analytics capabilities will, over time and with increasing datasets, feed into opportunities for lengthening revalidation and requalification windows and better alignment of maintenance models — away from preventative measures and toward predictive activities.
Conclusion
Modernizing cleaning validation with a fully digitalized approach is both a compliance necessity and a business opportunity. Intelligent, centralized platforms can eliminate bottlenecks, improve inspection readiness, and unlock operational agility.
Transitioning from paper-based methods to digital systems allows organizations to embed science- and risk-based practices throughout the validation lifecycle, while unburdening technical specialists through living, agile processes. This shift strengthens data integrity, ensures audit readiness, and provides the transparency and state of continuous control regulators expect. At the same time, automation reduces manual effort, shortens review cycles, and minimizes errors, freeing SMEs to focus on higher-value activities.
Scalability and standardization further extend these benefits. With digitalization, best practices can be deployed across global networks while preserving local flexibility where justified. Central oversight is always maintained, ensuring consistency without limiting site-level adaptability.
Looking forward, alignment with Pharma 4.0 and the integration of AI will enable predictive insights, smarter cleaning cycles, and continuous improvement. Digital cleaning validation will evolve alongside regulatory expectations, supporting long-term compliance and resilience.
Ultimately, digitalization repositions cleaning validation from a perceived burden into a strategic enabler of efficiency, competitiveness, increased output, and innovation across the life sciences industry. By adopting digital solutions today, organizations can safeguard compliance while preparing for the future of manufacturing.
Learn how ValGenesis can help modernize your cleaning validation program.
References
European Commission. (2011). EudraLex—Volume 4: EU guidelines for good manufacturing practice for medicinal products for human and veterinary use, Annex 11: Computerised systems.
https://health.ec.europa.eu/system/files/2016-11/annex11_01-2011_en_0.pdf
European Commission. (2015). EudraLex—Volume 4: EU guidelines for good manufacturing practice for medicinal products for human and veterinary use, Annex 15: Qualification and validation.
https://www.gmp-compliance.org/guidelines/gmp-guideline/eu-gmp-annex-15-qualification-and-validation
Food and Drug Administration. (1997). 21 CFR Part 11—Electronic records; electronic signatures.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-11
Food and Drug Administration. (2011). Process validation: General principles and practices.
https://www.fda.gov/media/71021/download
Food and Drug Administration. (2018). Data integrity and compliance with drug CGMP: Guidance for industry.
https://www.fda.gov/media/119267/download
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2005). ICH Q9: Quality risk management.
https://database.ich.org/sites/default/files/Q9_Guideline.pdf
International Society for Pharmaceutical Engineering. (2023). ISPE Baseline® Guide: Pharma 4.0™ (Vol. 8).
https://ispe.org/publications/guidance-documents/baseline-guide-vol-8-pharma-40-1st-edition
World Health Organization. (2014). WHO good manufacturing practices for pharmaceutical products: Main principles (WHO Technical Report Series No. 986, Annex 2).
https://www.who.int/publications/m/item/trs986-annex2