
In part 1 of this series, we explored why cleaning validation continues to top FDA inspection findings and why traditional compliance-only approaches often fall short. In part 2, we’ll examine the building blocks of a modern cleaning validation strategy, the factors that can disrupt a validated process, and the industry’s move toward risk-based, lifecycle-driven pharmaceutical cleaning validation.
Current Practices in Cleaning Validation
Cleaning validation in pharma has evolved from a box-checking compliance requirement into a scientifically rigorous, risk-based process that enhances both patient safety and manufacturing efficiency. Regulators now expect manufacturers to implement cleaning validation best practices that not only meet GMP standards but also integrate ongoing monitoring and continuous improvement.
This shift reflects a broader trend in pharmaceutical quality systems: moving from static validation exercises toward dynamic lifecycle management. By embedding risk management into every stage of cleaning validation, companies can ensure compliance while also improving efficiency and agility.
Read related post: What are the Regulatory Expectations for Cleaning Validation?
The Building Blocks of a Cleaning Validation Strategy
At the heart of modern pharmaceutical cleaning validation is a well-structured strategy that governs how cleaning processes are developed, tested, and maintained over time. Strong documentation is critical. Regulators expect to see a complete package that covers planning, execution, and review.
The typical components include a cleaning validation master plan (CVMP), which defines responsibilities, acceptance criteria determination methods, and overall strategy. Supporting this is a detailed cleaning validation protocol that describes how validation studies will be conducted, and a cleaning validation report that summarizes study results. Records and logs document every step of execution to ensure traceability for each cleaning run, while standard operating procedures (SOPs) guarantee consistency across teams. Analytical methods provide the scientific backbone, confirming that cleaning removes chemical, physical, and microbial contaminants to safe levels.
Together, these elements create a cleaning validation strategy that is transparent, reproducible, and inspection-ready.
Worst-Case Assessments: Preparing for the Toughest Scenarios
One of the most powerful tools in a cleaning validation strategy is the “worst-case” assessment. Manufacturers use this approach to design validation studies around the most challenging products and equipment, on the basis that if these pass, everything else will too.
For example, a company may select the most toxic or hardest-to-clean product in its portfolio to validate a multiproduct line. Similarly, the most complex or difficult piece of equipment is chosen to represent a broader equipment group. By targeting the extremes, companies can prove the adequacy of their cleaning validation process while optimizing time and resources.
Cleaning validation risk assessments add another layer of assurance by systematically identifying potential hazards and evaluating their impact. This structured approach balances efficiency with patient safety, reducing the chance of gaps in validation coverage.
Setting Limits and Acceptance Criteria
Effective cleaning validation requires clear limits for chemical, physical, and microbiological residues. These limits are typically calculated using a combination of approaches: sophisticated measures like Acceptable Daily Exposure (ADE) or Permitted Daily Exposure (PDE), or simpler thresholds like 10 parts per million (PPM) and toxicological values such as the No Observed Effect Level (NOEL). Regulators increasingly emphasize the use of ADE and PDE because they are science-based and product-specific.
The most conservative value is typically applied to define the maximum allowable carryover (MACO), which sets the benchmark for whether cleaning has been successful. Validation teams also establish acceptance criteria and sampling strategies — most commonly swab sampling and rinse sampling — to verify cleaning effectiveness. Careful selection of sampling locations is critical, focusing on areas that are hardest to clean or most likely to retain residues, such as valves, gaskets, or shadowed areas. Visual inspection is often performed as a supplementary check, but regulators caution that it should not serve as the sole acceptance criterion.
When to Revalidate: Triggers and Challenges
The best-designed cleaning validation programs are not static. Regular processes and events require companies to reassess and revalidate their processes. These include the introduction of new products with unique properties, changes to the equipment train, modifications to cleaning agents, or shifts in manufacturing processes. Deviations and failures are also important triggers, requiring investigation and corrective action.
When these situations arise, manufacturers must conduct thorough document reviews, perform impact assessments, and verify critical data such as MACO limits, equipment surface areas, and product batch sizes. This ensures that the validated state is maintained and that cleaning validation procedures continue to meet regulatory and safety requirements.
From Traditional Validation to Lifecycle Management
Traditional cleaning validation often focused narrowly on process qualification — proving once that equipment could be cleaned effectively — but failed to address ongoing performance or evolving risks. This static approach often led to inefficiencies, recurring failures, and weak post-validation monitoring.In contrast, regulators now emphasize a lifecycle model for cleaning validation. The FDA’s Process Validation Guidance (2011) introduced a three-stage framework that integrates risk management and continuous improvement. Other global authorities, including EMA and PIC/S, have echoed this shift, underscoring that validation is no longer a one-time exercise but a continuous process of control and verification.
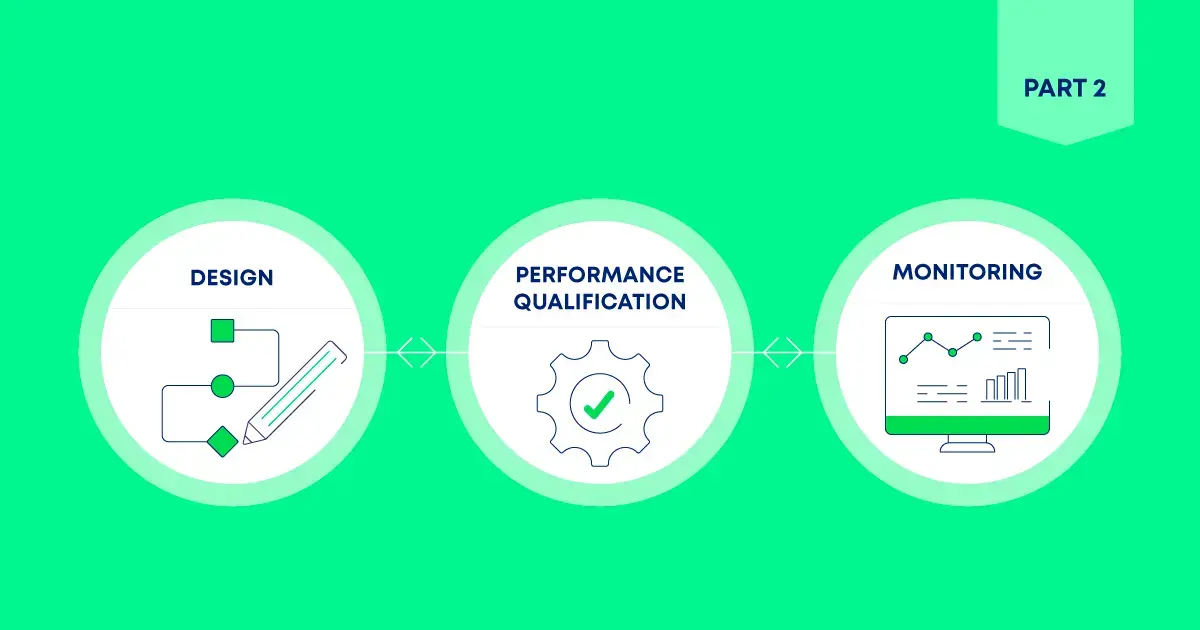
This lifecycle approach unfolds across three stages:
- Stage 1: Design — Developing process knowledge, evaluating risks, and defining control strategies.
- Stage 2: Performance Qualification — Testing under expected operating conditions to confirm that cleaning processes consistently meet acceptance criteria.
- Stage 3: Continued Process Monitoring — Ongoing monitoring and data analysis to ensure cleaning effectiveness over time, enabling a predictive quality mindset.
Figure 1 illustrates how cleaning validation maturity increases across these stages, with organizations evolving from basic compliance to proactive, data-driven oversight. Transitioning to a lifecycle approach requires ensuring the site is equipped with the right resources and technologies early in the development of cleaning procedures. Digital enablers, such as risk-based automated cleaning validation solutions, represent the next stage in the evolution of cleaning validation — a topic we’ll explore next.
![Figure-2-[BP]-The-Future-of-Cleaning-Validation-Part-2 (1)](https://www.valgenesis.com/hs-fs/hubfs/Figure-2-%5BBP%5D-The-Future-of-Cleaning-Validation-Part-2%20(1).png?width=1200&height=630&name=Figure-2-%5BBP%5D-The-Future-of-Cleaning-Validation-Part-2%20(1).png)
Figure 1: Cleaning validation level of maturity (Gorsky et al., 2017).
Looking Ahead to Part Three
The shift from compliance-based validation to a lifecycle-driven model marks a major milestone in how the industry approaches pharmaceutical cleaning validation. But the journey doesn’t stop there. In part 3, we’ll explore how digital technologies and Pharma 4.0 innovations are transforming cleaning validation even further — enabling real-time monitoring and predictive analytics that are redefining what compliance looks like in the future.
Ready to learn more? Watch the Video: How Digitalizing Cleaning Validation Leads to Shorter Timelines and Fewer Errors
References:
Food and Drug Administration. (2011). Process validation: General principles and practices (Guidance for Industry). U.S. Department of Health and Human Services. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/process-validation-general-principles-and-practices
Gorsky, I., Hanf, M., Hartman, C., & Long, M. (2017). How clean is clean in drug manufacturing: Cleaning validation level of maturity. American Pharmaceutical Review. https://www.americanpharmaceuticalreview.com/Featured-Articles/333832-How-Clean-is-Clean-in-Drug-Manufacturing-Cleaning-Validation-Level-of-Maturity/
Almeida, R. & Pierce, K. (2025). Industry insights: The future of cleaning validation. ValGenesis. https://www.valgenesis.com/resource/industry-insight-the-future-of-cleaning-validation
Table of Contents
Related Blog Posts


