
The concept of process validation has been evolving since 1978 when the FDA issued “good manufacturing practice (GMP) regulations.”
In the field of pharmaceutical manufacturing, process validation aims for excellence in product quality, safety, and efficacy. It is a systematic approach that goes beyond mere compliance, encompassing a series of stages to ensure that each step of the manufacturing process consistently produces a product that meets predefined specifications. Ultimately, it contributes to the delivery of pharmaceuticals that are safe, effective, and consistent in their therapeutic outcomes.
According to the FDA, process validation is the systematic collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering a quality product (FDA, Guidance for Industry – Process Validation: General Principles and Practices, 2011). As depicted in Figure 1, the suggested workflow encompasses three stages: process design (stage 1), process qualification (stage 2), and continued process verification (stage 3).
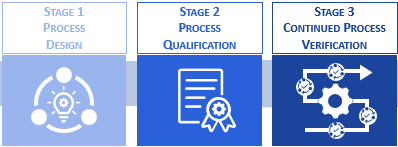
Figure 1: The three stages of process validation according to the FDA.
Other agencies such as EMA have been releasing guidelines on process validation, for example, the 2017 EMA Guideline on Process Validation for Finished Products, which are later adopted by each country and become legally binding requirements.
Both agencies, the FDA and EMA, are generally aligned in the concept that validation is a multi-stage lifecycle, from process design to performance qualification and commercial manufacturing, where product and process knowledge is incrementally gained. Organizations that join regulators and industry, such as ICH, have been contributing to the topic of process validation for over a decade.
Furthermore, according to the EMA Guideline on Process Validation for Finished Products and GMP Annex 15 (Qualification and Validation), process validation can be performed in two fashions: traditional process validation, continuous process verification, or a combination of both.
Two Ways to Perform Process Validation
Traditional process validation is typically applied following pharmaceutical or process development, after the scale-up to commercial production, and prior to marketing the finished product. In the traditional approach, several batches of the finished product are manufactured under routine conditions to confirm that the process is reproducible and that product quality is consistent.
Continuous process verification is an alternative approach to traditional process validation in which manufacturing process performance is continuously monitored and evaluated (ICH Q8 – Pharmaceutical Development). Continuous process verification is a science and risk-based real-time approach to verify and demonstrate that a process that operates within the predefined specified parameters consistently produces material that meets all its critical quality attributes (CQAs) and control strategy requirements.
Deciding which approach to use will depend on having sufficient knowledge and understanding of the process, which in turn depends on several factors, such as:
- Prior development and manufacturing knowledge from similar products and/or processes.
- The extent of process understanding gained from development studies and commercial manufacturing experience.
- The complexity of the product and/or manufacturing process.
- The level of process automation and analytical technologies used.
Additionally, to enable continuous process verification, you must:
- Perform extensive in-line, on-line, or at-line controls and monitor the process performance and product quality of each batch.
- Collect relevant data on the quality attributes of incoming materials or components, in-process material, and finished products, including attributes, parameters, and end points, and an assessment of CQA and critical process parameter (CPP) trends.
- Employ process analytical technology (PAT) applications such as NIR spectroscopy with or without a feedback control loop and multivariate statistical process control (MSPC).
Common Aspects of Process Validation in the Pharmaceutical Industry
Independently of whether a medicinal product is developed by a traditional or enhanced approach, the manufacturing process must be validated before the product is placed on the market. This requires the collection and evaluation of data from the process design stage through commercial production to establish scientific evidence that a process is capable of consistently delivering quality products.
The validation process should:
-
Confirm that the control strategy is adequate for the process design and the quality of the product.
- Involve a series of activities that occur throughout the lifecycle of the product and process.
- Cover all manufactured strengths and all manufacturing sites used to produce the marketed product.
- Generate process validation data to demonstrate the adequacy of the manufacturing process at each site of manufacture.
- Be carried out in accordance with GMP guidelines, and data should be stored at the manufacturing location, making it readily accessible for inspection purposes.
- Include the application of a quality risk management approach with clear documentation on how the risk assessments are used to support the validation activities.
Best Practices for Process Validation
Process validation should be viewed as an ongoing and dynamic process that ensures the manufacturing process remains effective, efficient, and aligned with evolving regulatory standards throughout the entire product lifecycle. As such, process validation should cover all intended marketed strengths and sites of manufacture.
Regularly revisiting and reassessing validation protocols allows organizations to identify areas that can be refined, optimized, or strengthened.
Process Validation Protocols
Process Validation protocols should define the critical process parameters (CPPs), the critical quality attributes (CQAs), and the related acceptance criteria. It should include:
- A description of the process and a reference to the master batch record.
- Functions and responsibilities.
- A summary of the CQAs to be considered and specification limits.
- A summary of the CPPs and their limits.
- A summary of other attributes and parameters to be investigated and monitored, as well as reasons for their inclusion.
- A list of equipment and facilities to be used along with their calibration status.
- A list of analytical methods and method validation.
- In-process controls with acceptance criteria.
- Additional testing to be carried out.
- A sampling plan.
- Methods for registration and evaluation of results.
- The process for batch release and certification.
Applying Quality Risk Management to Process Validation
The FDA emphasizes the importance of employing risk-based decision-making throughout the lifecycle of process validation.
By implementing a quality risk management (QRM) system, pharmaceutical companies can identify, assess, and mitigate potential risks throughout the product lifecycle.
Risk assessment methodologies ensure that the manufacturer’s efforts are focused on the areas of highest risk by addressing critical process parameters and potential failure modes. This transforms QRM into a proactive tool when integrated into process validation.
This risk-based approach not only enhances the efficiency of validation activities but also reinforces the adaptability of processes in the face of changing conditions. All attributes and parameters are evaluated in terms of their roles in the process and their impact on the final product or intermediate materials and reevaluated as new information becomes available. The degree of necessary control over those attributes or parameters is proportional to their risk to the process and process output.
The application of QRM to process validation is not just a regulatory expectation but a fundamental strategy for ensuring the ongoing quality, safety, and efficacy of pharmaceutical products.
Quality Attributes and Process Parameters
As established, the process validation protocol should define whether all quality attributes and process parameters, which are considered important for ensuring the validated state and acceptable product quality, can be consistently met by the process.The basis by which process parameters and quality attributes are identified as being critical or non-critical should be clearly documented, taking into account the results of the risk assessment activities.
Adopting a lifecycle approach to process validation by employing risk-based decision-making throughout that lifecycle improves the usefulness of criticality interpretation by turning it into a continuum rather than a one-off exercise.
Validation Batches
Homogeneity within a batch and consistency between batches are goals of process validation activities.Batches manufactured for process validation should be the same size as the intended commercial-scale batches. Any use of different batch sizes must be justified. Batches should only be manufactured by trained personnel in accordance with GMP guidelines using approved documentation.
Digitalization of Process Validation
Validation is a crucial step in the manufacturing process, yet many companies still rely on manual methods despite the high costs, frequent human errors, and inefficiencies that come with it. This is a barrier to the implementation of dynamic process validation. By incorporating the latest technologies and methodologies, companies can streamline these processes and enhance the overall quality of pharmaceutical products.
The bottom line is that life sciences manufacturers should digitalize their validation operations. Moreover, regulatory authorities have been encouraging the industry to embrace digital tools to manage the entire validation lifecycle.
Taking the Digital Approach Toward Continued Process Verification
As previously stated, according to the FDA terminology, the goal of the third validation stage (continued process verification) is the continual assurance that the process remains in a state of control (the validated state) during commercial manufacture. This is done by collecting and analyzing product and process data that relate to product quality.The most effective way to perform this is to:
- Integrate all the systems for registry, collection, and storage of data.
- Be able to check process status in real-time through monitoring dashboards.
- Accelerate and automate report generation without human input.
ValGenesis is a pioneer in this field with the development of iCPV.
The Business Case for Digitizing Your Validation Lifecycle
Manual validation is prone to human error. It stifles innovation, increases compliance risk, and hinders time to market. These inefficiencies cost regulated companies tens of millions of dollars annually.
Companies that still do not use paperless validation software face significant challenges: the high costs associated with risk management, validation, and the subsequent change management and continued qualification to maintain the validation status throughout the lifecycle of entities.
ValGenesis iVal is designed to manage the entire validation lifecycle electronically and remove the inefficiencies that plague paper-based processes. By implementing ValGenesis iVal, you can expect efficiency improvements of up to 50%.
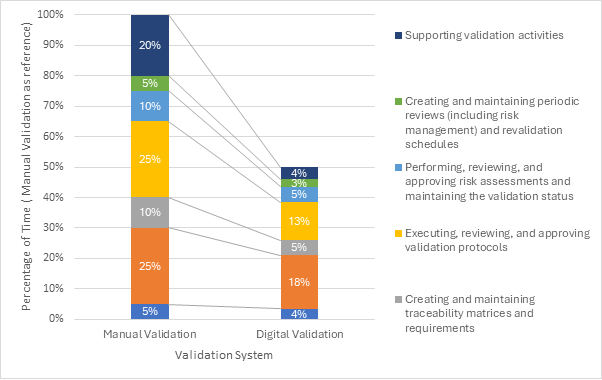
Figure 2: The impact of digital validation on validation operations.
To learn more, contact one of our validation experts.
Table of Contents
Related Blog Posts

.webp)
.webp)