This blog is the first of a three-part series focused on FDA’s Computer Software Assurance (CSA). It explains how to accelerate CSA adoption within your organization.
What Exactly is CSA?
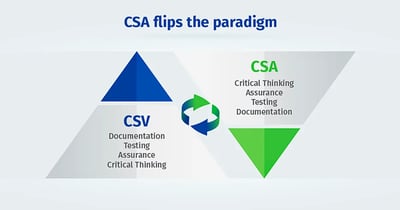
There are two primary things CSA delivers. First, CSA flips the traditional validation framework (CSV) upside down to a new way of thinking that’s right side up. Second, CSA charts a path through a critical thinking process to ensure appropriate, risk-based, least burdensome validation.
Flipping the Paradigm Right Side up
With traditional CSV, too much focus is on documentation. The amount of time and effort put into documentation consumes significant resources and sacrifices critical thinking. CSA inverted this upside-down way of thinking, placing the primary focus on critical thinking, then assurance needs, then finally, testing and documentation. This paradigm shift is illustrated in figures one and two below.
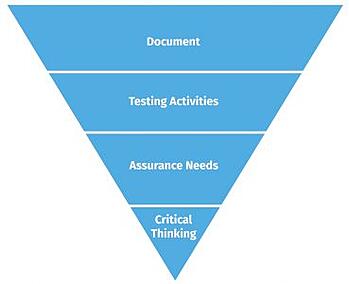
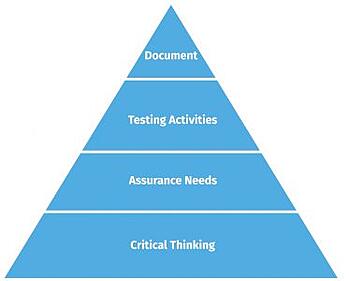
Figure One - The old CSV way Figure Two - The new CSA way
Charting a CSA Path Through a Critical Thinking Process
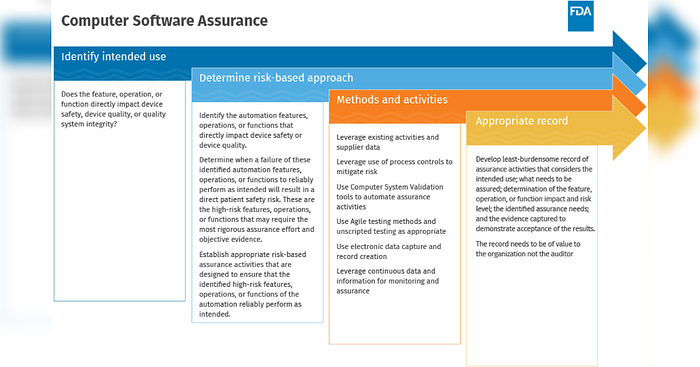
Figure Three - How to CSA (SOURCE: Cisco Vincenty, FDA Case for Quality Program Manager (FDA))
Identify Intended Use
The new CSA approach starts with identifying intended use. Here it’s determined if a system, feature, operation, or function directly impacts device safety, device quality, or quality system integrity. How ValGenesis supports CSA: ValGenesis’ VLMS accelerates critical thinking using the system assessment functionality to identify the intended use.
Determine Risk-based Approach
The next step in the computer software assurance process is to determine a risk-based approach, focusing on safety and quality, specifically high-risk areas that may require the most rigorous assurance effort and objective evidence, and ensure the high-risk features, functions, or operations reliably perform as intended. How ValGenesis supports CSA: ValGenesis’ VLMS accelerates risk assessments with risk assessment functionality at the system level and requirement level.
Methods and Activities
Where CSA's value shines bright is leveraging vendor documentation, ad-hoc testing, and automated testing; this is a risk-based, least burdensome approach. The biggest payoff is with medium- and low-risk features where little or no rigorous effort is required. Validation cycles are further accelerated by validation leveraging automation, iterative methodologies (e.g., Agile), as well as continuous monitoring and verification. In other words, digitize validation and go paperless. How ValGenesis supports CSA: ValGenesis’ VLMS accelerates methods and activities using patented requirements traceability matrices (RTMs), frameworks, and test functions.
Appropriate Record
Complete validation with a high degree of rigor is necessary for high-risk systems. For medium-risk systems, less rigor can be applied. For low-risk systems, little or no validation may be required. If it is, in fact, low, there should be no impact in the event of failure. If there is a negative consequence for a low-risk failure, then the risk assessment was inaccurate, or the process is flawed. How ValGenesis supports CSA: ValGenesis' VLMS accelerates appropriate records with electronic test case execution and deviation management to create and maintain validated states and records.
Change Control
Change control is necessary to ensure changes to features, functions, or operations follow the CSA process. This maintains the system in its appropriate state (e.g., “validated”). How ValGenesis supports CSA: ValGenesis' VLMS accelerates appropriate record change control through change management and periodic scheduling to maintain an appropriate validated state on a continuous basis.
Change the Culture and Process by Igniting Teams
Culture must change from a compliance-centric mindset to quality-focused culture. This is discussed in more detail in part two.
Related content: Your Guide to Computer Software Assurance
Resources:
(1) FDA Computer System and Software Validation – What You've Known for 20+ Years is Changing, Jon Speer, Greenlight Guru, December 2, 2018.